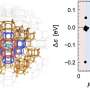
A research team has introduced a groundbreaking design principle for QM/MM (quantum mechanics/molecular mechanics) simulations that promises to significantly enhance the accuracy and efficiency of multiscale molecular modeling. This new approach facilitates the automatic and objective identification of the quantum-mechanical region based on changes in electronic states, providing a solution to a long-standing challenge in the field.
The development comes at a time when the demand for precise molecular simulations is increasing across various scientific domains, including materials science, biochemistry, and drug discovery. Traditional methods have often struggled with the ambiguity in defining the quantum region, which is crucial for accurate results. By focusing on the electronic-state responses, this new principle aims to streamline the simulation process, making it more reliable and user-friendly.
Enhancing Multiscale Molecular Simulations
The innovative design principle offers a significant advancement in how researchers can approach complex molecular systems. Previous QM/MM methods frequently required manual intervention to delineate the quantum region, which could lead to inconsistencies and errors. With the new technique, the research team proposes that the identification of quantum mechanics regions can be performed automatically, thereby minimizing human error and enhancing reproducibility.
This development is particularly noteworthy given the increasing complexity of molecular systems being studied today. According to the research team, addressing the challenges associated with electronic-state changes not only simplifies the simulation process but also opens up new avenues for research. The implications are vast, impacting fields such as drug design, where accurate molecular interactions can lead to more effective therapies.
Future Implications and Applications
The potential applications of this new QM/MM design principle extend beyond just theoretical interest. Its implementation could lead to more accurate predictive models in various scientific fields, allowing researchers to simulate chemical reactions and molecular interactions with unprecedented precision. The research team anticipates that the integration of this approach into existing simulation software could revolutionize how scientists conduct their work.
As the scientific community continues to seek innovative solutions for complex problems, this new design principle stands out as a promising advancement. The research team emphasizes the importance of collaboration across disciplines to further refine and implement these findings. With ongoing advancements in computational power and algorithms, the future of molecular simulations looks increasingly promising.
Overall, this development marks a significant step forward in the evolution of QM/MM simulations, promising to enhance both the accuracy and efficiency of molecular modeling for researchers worldwide.